



2022 kavli prize in Neuroscience
2022 Kavli
Prize in
Neuroscience
The Norwegian Academy of Science and Letters has decided to award the 2022 Kavli Prize in Neuroscience to Jean-Louis Mandel, Harry T. Orr, Christopher A. Walsh and Huda Y. Zoghbi
Jean-Louis Mandel
University of Strasbourg, France
Read the full bio
Harry T. Orr
University of Minnesota Medical School, USA
Read the full bio
Christopher A. Walsh
Howard Hughes Medical Institute, Harvard Medical School, Boston Children's Hospital, USA
Read the full bio
Huda Y. Zoghbi
Howard Hughes Medical Institute, Baylor College of Medicine, Texas Children´s Hospital, USA and Lebanon
Read the full bio




"for pioneering the discovery of genes underlying a range of serious brain disorders"
Committee Members
- Kristine B. Walhovd (Chair), University of Oslo, Norway
- Mary E. Hatten, The Rockefeller University, USA
- Angela Friederici, Max Planck Institute for Human Cognitive and Brain Sciences, Germany
- John O’Keefe, University College London, UK
- Antoine Triller, Institut de Biologie de l'École Normale Supérieure, France
Citation from the Committee
Understanding inherited brain disorders has been made possible by the novel genetic approaches developed by the current laureates, leading to improved care for patients and their families. The laureates identified genes underlying brain diseases and developed novel strategies for diagnosis and treatment: Mandel for fragile X syndrome; Zoghbi and Orr for spinocerebellar ataxia type 1; Zogbhi for Rett syndrome; and Walsh for some epilepsies and developmental brain disorders.
Jean-Louis Mandel is a key contributor to the discovery that a repeated genetic sequence accounts for the pattern of inheritance of the Fragile X syndrome, a major cause of intellectual disability. Mandel showed that the expression of the Fragile X syndrome depended on methylation of certain sites. His work led to the development of widely used diagnostic tools. Mandel also characterized the FMRP protein that binds mRNA and affects the levels of protein expressed in neurons. Understanding unstable repeat expansions provided a model for numerous neurological diseases.
Huda Zoghbi and Harry Orr independently discovered ATAXIN1, the gene underlying the neurodegenerative disease spinocerebellar ataxia type 1 (SCA1) that causes loss of balance and coordination. The ATAXIN1 mutation is a repetition of a CAG trinucleotide sequence in the gene, with the size of the repeat inversely correlated with the age of onset of the disease. They also discovered that the ataxin-1 encodes a DNA binding protein that causes the formation of large protein aggregates in cerebellar neurons, leading to their degeneration. Using oligonucleotide therapy, they showed improvement of symptoms in a mouse model. Zoghbi also discovered the mutations in the MECP2 gene that cause Rett syndrome, which develops in young girls with devastating motor and cognitive symptoms. She showed that changes in the level of MECP2, a repressor of gene expression is essential for the normal function of many types of neurons in the brain. MECP2 is one of the first identified epigenetic causes for a brain disorder. Using oligonucleotide therapy, she found that normalizing MeCP2 levels reverses the effects of MECP2 duplication in mice.
Christopher Walsh identified genetic mutations that underlie disorders affecting the cerebral cortex, including adverse sporadic and somatic cell mutations in the developing brain. Many of these discoveries came from one of Walsh’s key innovations to study recessive mutations in geographically isolated families. These mutations caused some forms of epilepsy and autism spectrum disorders.

The Huda Zoghbi Lab - The laboratory uses genetic, biochemical, and cell biological approaches to explore the pathogenesis of polyglutamine neurodegenerative diseases and Rett syndrome, and to study genes essential for normal neurodevelopment.
Revealing the Genetic Surprises at the Roots of neurological disease
Before scientists mapped the human genome in the early 2000s, the quest for the genes at the roots of disease was a painstaking and sometimes baffling process. Yet, the four scientists honored with the 2022 Kavli Prize in Neuroscience persevered, eventually “pioneering the discovery of genes underlying a range of serious brain disorders.”
By Lindsay Borthwick, science writer
Before scientists mapped the human genome in the early 2000s, the quest for the genes at the roots of disease was a painstaking and sometimes baffling process. Yet, the four scientists honored with the 2022 Kavli Prize in Neuroscience persevered, eventually “pioneering the discovery of genes underlying a range of serious brain disorders.”
Each discovery tells a story about where neurological disorders begin within the vastness of the genome and how they develop — from gene to protein to disease. By uncovering these pathways, Jean-Louis Mandel, Harry Orr, Huda Zoghbi and Christopher Walsh have established a blueprint for neuroscience research and pushed our understanding of the nervous system in unexpected directions.
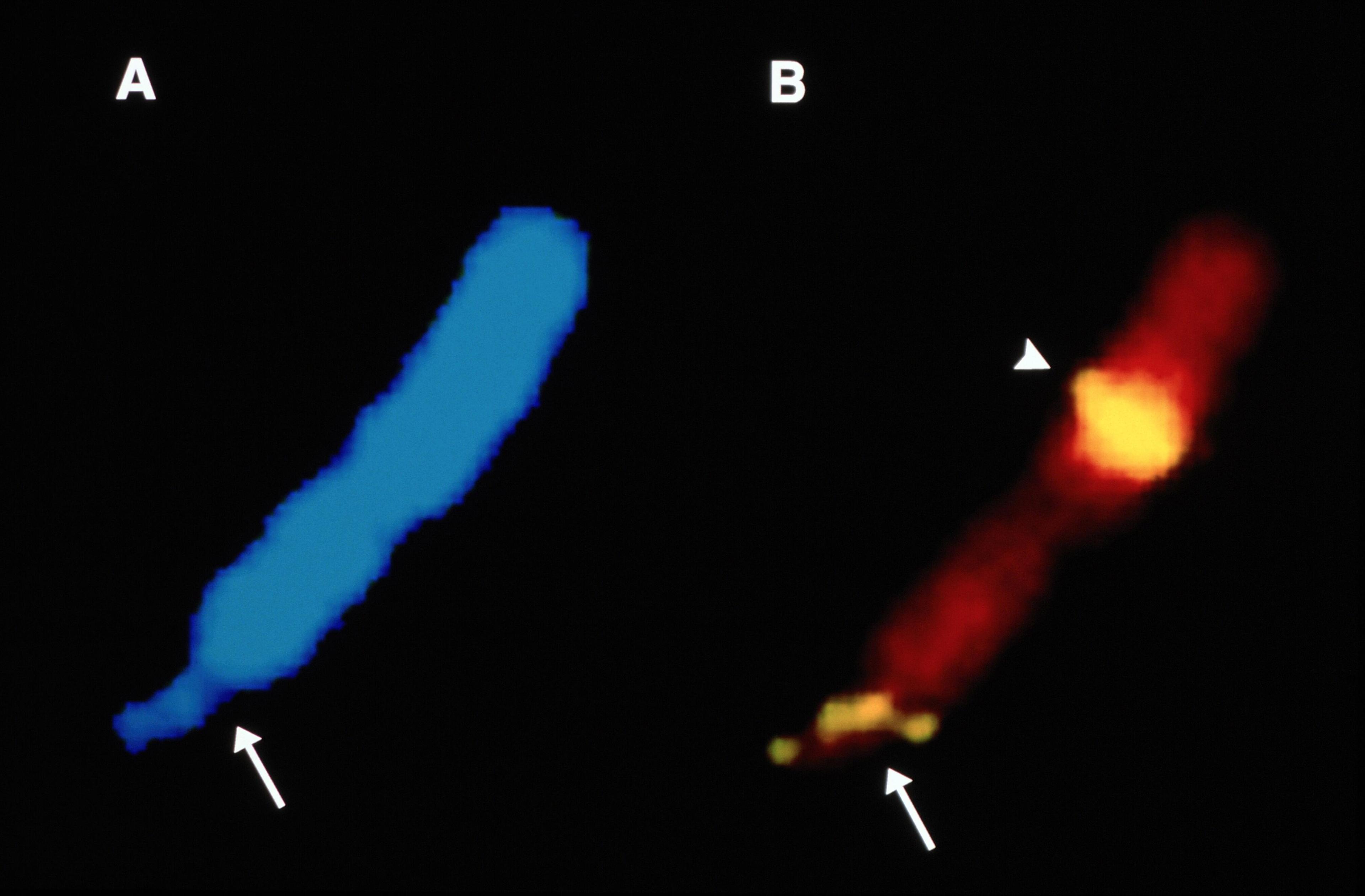
Mandel began his search on the X chromosome, a hoped-for shortcut to the mutations that cause hereditary diseases. Eight years later, in 1991, he discovered an unusual mutation in a gene on the X chromosome that caused Fragile X syndrome, an inherited form of intellectual disability and autism.

Fragile X syndrome is caused by an unusual mutation on the FMR1 gene. The mutation consists of a three-letter DNA repeat that causes structural changes, toward the end of X chromosome, that can be seen with the naked eye. The arrows mark the fragile sites on the chromosomes. Credit: Dr. Ben Oostra (CC0 1.0 Universal)
The mutation wasn’t a simple change to a letter in the DNA sequence. Mandel showed that the mutation was a string of triple-letter repeats that disrupted the FMR1 gene. Such repeats result in the loss of the FMRP protein, which affects higher brain function. One of Mandel’s most unique contributions is that he showed that the expression of the Fragile X syndrome depends on epigenetic modificiations epigenetic modification at certain sites in the FMR1 gene. Mandel´s work also led to improvements in diagnosis.


It is now recognized that three-letter repeats of DNA, like the one shown here, are involved in more than 50 hereditary diseases, including Fragile X syndrome, Spinocerebellar ataxia 1, and Huntington’s disease. As the number of repeats increases from generation to generation, disease symptoms develop earlier and are more severe. The TGC repeat shown here in red is associated with a disease of the cornea and blurred vision. Mandel showed that a string of triple-letter repeats — CGG, CGG, CGG, and so on — disrupted the FMR1 gene. People with Fragile X have at least 200 repeats in the gene and cannot make the FMRP protein, which is vital for brain function.Credit: Wieben, E.D. et al. (2012)
Today, “unstable repeat expansions” are recognized as a common disease mechanism responsible for more than 50 genetic disorders. A familiar pattern has also emerged: as the number of repeats increases with each generation, symptoms arise earlier and are more severe.
Similar repeat expansion
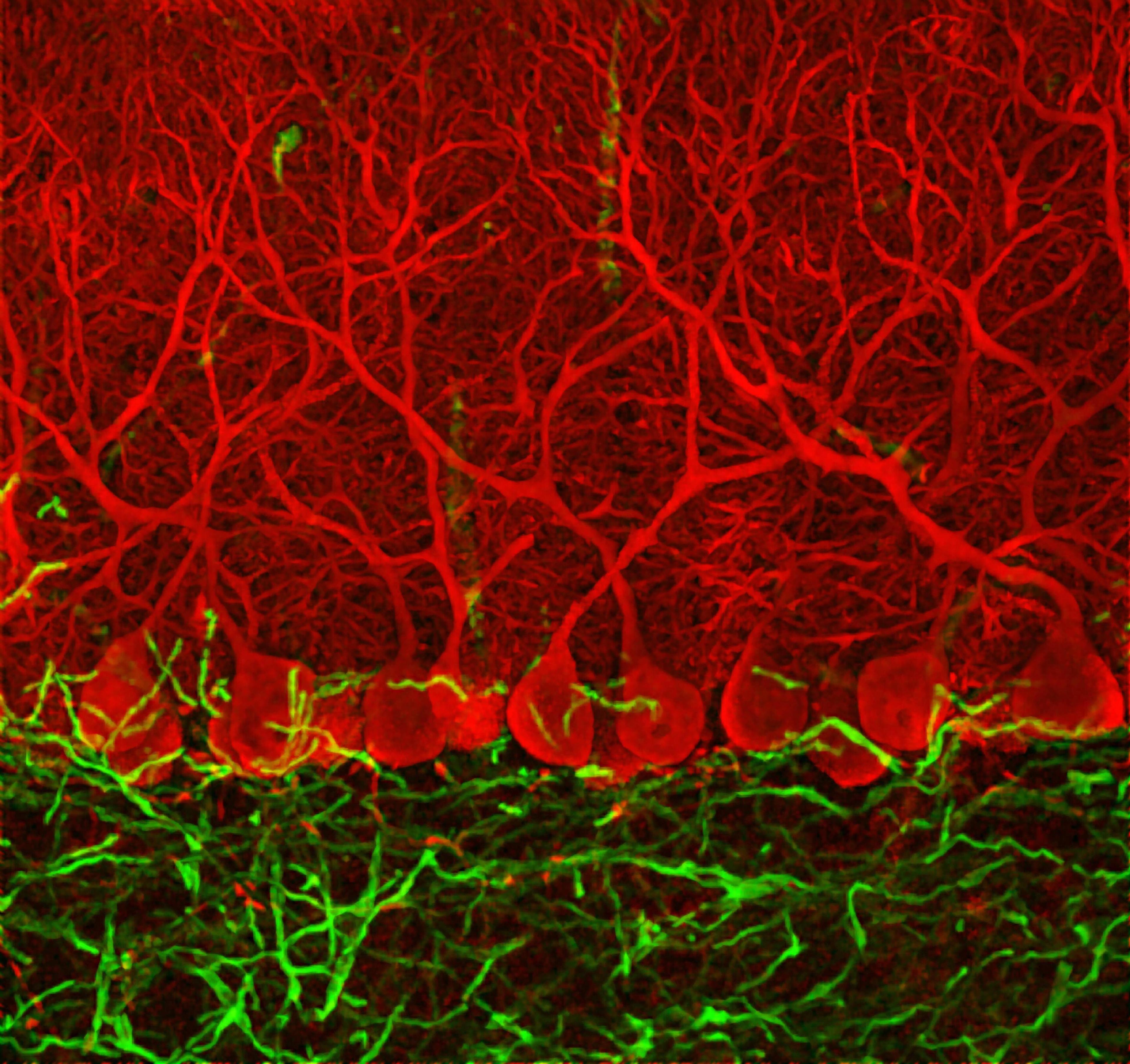
One of those other disorders is Spinocerebellar ataxia 1 (SCA1), in which neurons in the cerebellum degenerate. Orr and Zoghbi showed that a similar repeat expansion causes SCA1.

Purkinje cells (red) of the cerebellum, a brain region involved in voluntary movement, are easy to recognize because of their elaborate branches. The cells degenerate in spinocerebellar ataxia 1 (SCA1), an inherited disease caused by mutations in the ATXN1 gene. Harry Orr and Huda Zoghbi independently discovered the gene in 1993. As collaborators, they have worked out how the mutations lead to the degeneration of Purkinje cells in people with SCA1. Credit: Ludovic Collin. Attribution-NonCommercial 4.0 International (CC BY-NC 4.0)
Remarkably, on the same day in 1993, the two researchers independently discovered ATXN1, the gene responsible for SCA1. Then, working together, they pinned down the mechanism: the mutation causes proteins to misfold and clump together in cerebellar neurons, eventually leading to their death.
“The discovery [of repeat expansions] raised great interest in human genetics, and particularly in neurology, because these were important diseases for which people had no clues about their causes,” says Mandel.
Zoghbi didn’t stop at SCA1. From the outset of her career, she wanted to solve the mystery of Rett syndrome, an autism-like, rare genetic neurological disorder in which young girls progressively lose motor skills and language. In 1983, she was the first neurologist in the United States to diagnose two children with the disease.
Unlike Fragile X syndrome and SCA1, Rett syndrome isn’t hereditary, making it difficult to pinpoint the gene responsible. Eventually, her persistence paid off. Zoghbi found the gene, MECP2, that causes Rett syndrome in 2000, more than 15 years after her hunt began.
It was exciting for several reasons. First, MECP2 turned out to be a master regulator — a gene that controls the function of thousands of other genes. Too much or too little disrupts how the brain works. Second, the discovery that spontaneous mutations in MECP2 cause Rett syndrome changed the way researchers thought about autism — one of the most prevalent neurodevelopmental disorders.
“People immediately shifted and started sequencing individuals with autism. It really opened up discovery,” says Zoghbi.
Like Zoghbi, Christopher Walsh is also a neurologist. He has spent his career studying human brain development and how genetic mutations change the brain’s structure and function. His focus is the cerebral cortex, the region associated with cognition.
Walsh has discovered more than three dozen neurological disease genes. Mutations in some of these genes cause structural malformations that range from subtle to profound, including some that cause severe forms of epilepsy in children. In a few of those children, Walsh made another intriguing discovery: mutations that were present in some but not all the cells of the body.

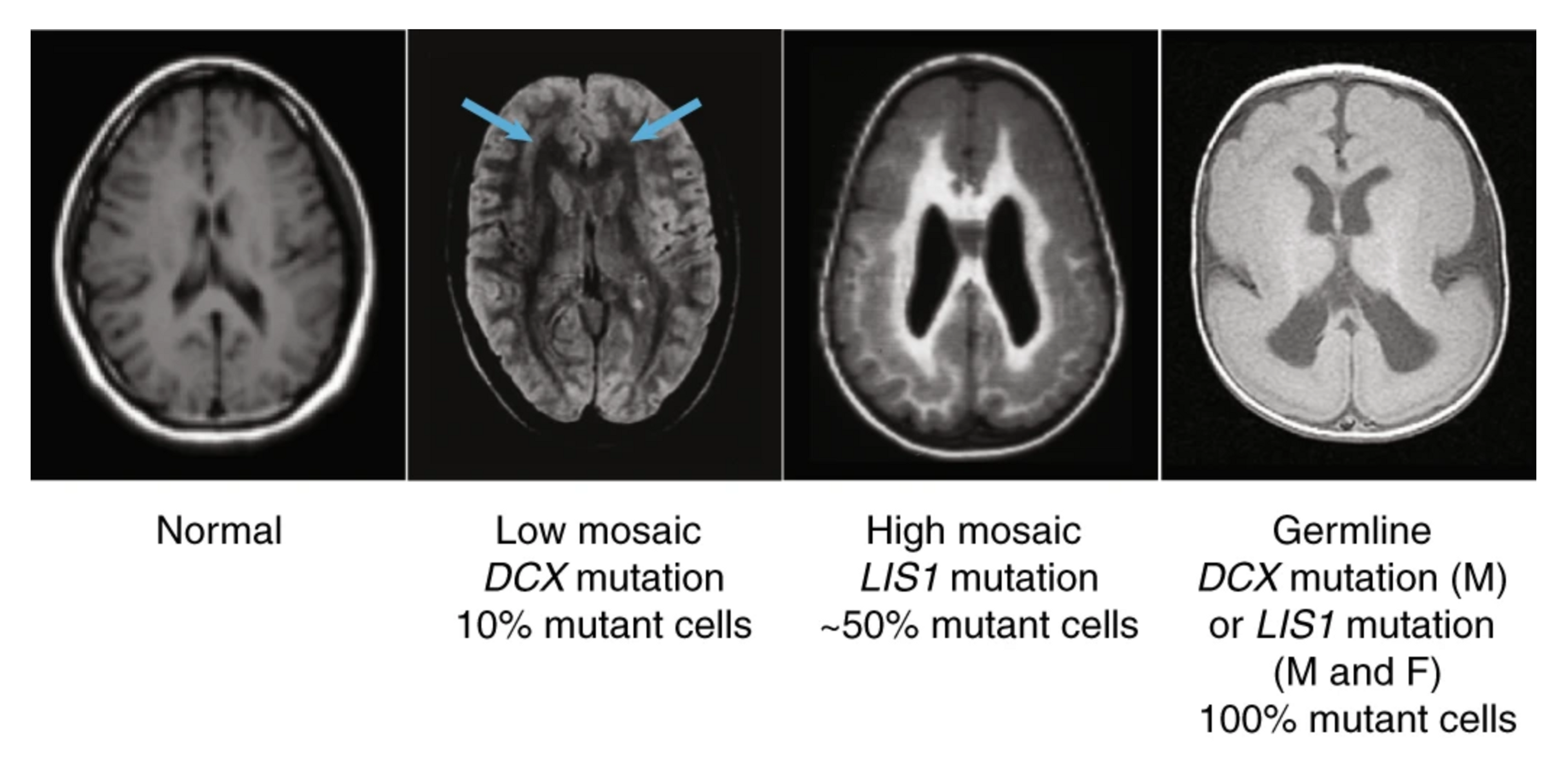
Christopher Walsh has discovered numerous genes that help guide the proliferation and migration of neurons during brain development, ultimately shaping the brain’s structure and function. Mutations in two of these genes, Double Cortin X (DCX) and Lissencephaly 1 (LIS1), cause rare structural malformations of the cerebral cortex that can be seen in these brain scans. Walsh discovered that these mutations are not inherited from parents but instead arise spontaneously with each generation. Credit: Springer Nature
The genetic changes, called somatic mutations, accumulate slowly, during development, at a rate of about 15 to 20 per year. Elderly neurons can have more than a thousand. Most of the mutations sit silently in the genome and don’t cause problems. But Walsh has shown that some disrupt genes and increase the risk of epilepsy, autism, and some neuropsychiatric disorders.
Somatic mutations represent a new genetic mechanism for brain disease. Their existance also means that every human brain is a mosaic, made up of billions of individual cells with slightly different genomes. As Walsh writes in his autobiography, “Every human is an experiment of nature.”
The laureates’ discoveries have helped illuminate the biology of the nervous system and moved the field toward therapies for disorders once considered untreatable. They have also provided a paradigm for discovery in neuroscience and beyond that will resonate for decades to come.